
Metabolomics Methods
What is metabolomics analysis:
A metabolomic analysis combines a number of steps from tissue extraction through the detection and measurement of metabolites to analysis, interpretation and visualization of the results.
All steps may be either untargeted or more targeted, the choice depending on the analytical/biological questions posed and various other factors, including the classes, stability and other properties of metabolites of interest, whether or not the samples need to be purified, detection limits and required analytical accuracy.
To be able analyze metabolites from a complex sample, e.g. plasma or tissue, they must to be isolated via extraction. This usually involves organic solvent such as methanol, acetonitrile or chloroform, depending compound class to analyze, and homogenization (e.g. in bead mill).
Stable isotope labelled standards are added to the extraction solvent to compensate for variation in the analytical procedure and check the quality of the analysis. Purification/fractionation can be performed if needed, for example, to improve sensitivity and for targeted analysis of specific compounds.
The extraction method in metabolomics analysis should:
- Extract the largest number of metabolites as unbiased as possible, independent on physical or chemical properties of the metabolites
- The extraction method should not cause degradation of metabolites
- The extraction must be reproducible
A metabolomic analysis combines a number of steps from tissue extraction through the detection and measurement of metabolites to analysis, interpretation and visualization of the results.
All steps may be either untargeted or more targeted, the choice depending on the analytical/biological questions posed and various other factors, including the classes, stability and other properties of metabolites of interest, whether or not the samples need to be purified, detection limits and required analytical accuracy.
More than 2000 putative metabolites can be detected using a combination of analytical techniques such combined gas chromatography – mass spectrometry (GC-MS) or liquid chromatography – mass spectrometry (LC-MS, LC-MSMS). However, only a fraction of the detected compounds will be annotated. The annotation is based on our in-house mass spectra libraries (GC-MS and LC-MSMS), as well as commercial and freely available libraries.
Depending on research question, we use one or several methods for analyzing the metabolome.
With targeted metabolite profiling, we analyse specific compound classes, e.g. amino acids, fatty acids, eicosanoids and steroid hormones. The advantage compared to an untargeted metabolomics approach is that with stable isotope labelled internal standards and calibration curves, absolute quantitative data can be achieved. Furthermore, depending on methods used, the sensitivity and specificity of the analysis is better.
The most common approach is LC-TQMS (triple-quadrupole mass spectrometry) in multiple-reaction-monitoring (MRM) mode for targeted profiling. Figure 3 shows example of analysis of short-chained fatty acids by LC-TQMS.
Examples of targeted methods available at the platform (type of mass spectrometer in “()”):
- TCA metabolites (GC-MS; LC-MS)
- Short-chained fatty acids (LC-MS)
- Hydroxylated fatty acids (LC-MS)
- Amino acids (LC-MS)
- Dipeptides (LC-MS)
- Bile acids (LC-MS)
- NAD-metabolites (LC-MS)
- Nucleotides, e.g. ATP, ZTP etc. (LC-MS)
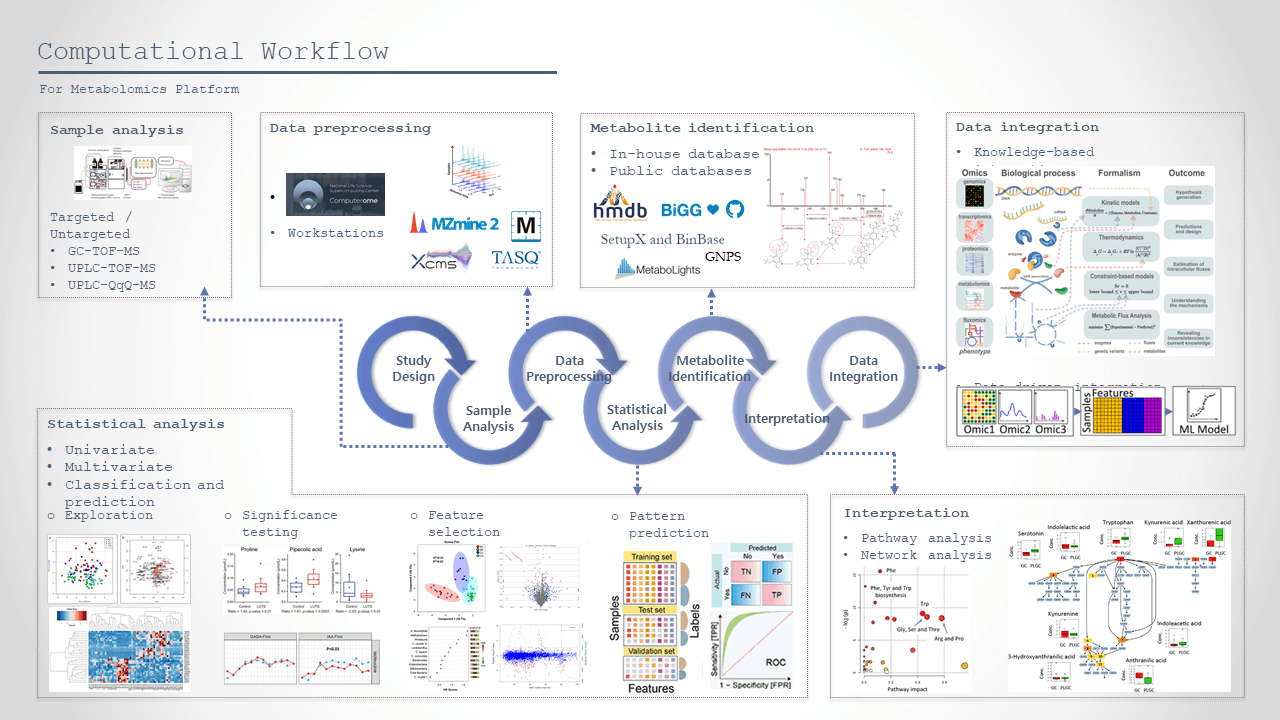
Metabolomics analysis demands an e3fficient computational workflow from mass spectrometry analysis to final statically analysis and interpretation. This involves e.g. data preprocessing of mass spectrometry data files, i.e. generating a table with detected mass features (putative metabolites with retention time and mass to charge ratio), annotation (identification) of mass features, statistical analysis, modelling, integration with other data and final interpretation.
Current computational workflow at the platform can be seen here.
{kind=link}